医療機器に該当することが確認できたら、次は「何という医療機器か(一般的名称)」「どのリスクランクに相当するか(クラス分類)」「どの手続きで上市できるか(申請区分)」を整理します。
これらは申請方針、必要試験の範囲、審査機関、審査期間の見通しに直結します。
Contents
なぜこれらを調べる必要があるのか
自社の開発機器がどの一般的名称・分類・区分に該当するかを把握することで、以下が明確になります。
- クラス分類(クラスⅠ〜Ⅳ)や申請区分が判定でき、必要な規制手続きや審査経路が決まる
- 提出書類・試験要件など開発計画に直結する要件が把握できる
- 保険適用や販売戦略を立てる際の基礎情報になる
早い段階で確認しておくことで、余計な手戻りや開発遅延を防ぐことができます。
一般的名称
一般的名称とは
医療機器には、それぞれの製品を大まかに分類・識別するための「一般的名称(Generic Name)」が定められています。これは、特定の製品名ではなく、機能や構造、使用目的など共通する特徴に基づいて付けられた名称で、規制・申請・保険適用などの基礎情報として活用されます。
一般的名称の調査方法
「医療機器該当性とは?調査方法を含めて解説」でも使用した、PMDAの一般名称検索を使うことで、既製品の医療機器情報(医薬品、再生医療を含む)を検索することができます。
- PMDA(医薬品医療機器総合機構)の「医療機器等基準関連情報 一般的名称検索」
- 同種同効の既承認機器の情報(承認書・届出書など)を参考にする
クラス分類とは
医療機器は、生体への影響の程度に応じて「クラスⅠ」「クラスⅡ」「クラスⅢ」「クラスⅣ」に区分されています。
このクラス分類によって、その後の開発プロセスが大きく異なるため、自社の開発機器がどのクラスに該当するかを早めに確認することが重要です。
GHTFルールに基づくクラス分類調査方法
GHTFルールとは
GHTF(Global Harmonization Task Force)は、医療機器の国際的な規制整合を目指して設立された組織で、医療機器のリスクベース分類(クラスA〜D)について基本的な考え方とルールを示しています。現在はIMDRF(International Medical Device Regulators Forum)に引き継がれ、各国規制当局の分類基準のベースになっています。
なぜGHTFルールが重要か
- 新しい市場(海外含む)に参入する際の共通言語になる
- 国内外の薬事戦略やクラス分類判定の裏付けに使える
- 開発初期から適切な試験・申請準備ができる
調査・判定の進め方
- 製品の基本情報を整理
使用目的、使用部位、侵襲性、体内留置時間、能動・非能動など、GHTFが分類で重視する要素を洗い出します。 - GHTFの分類ルールに当てはめる
GHTFの文書には「非侵襲型医療機器」「侵襲型医療機器」「能動型医療機器」「体外診断用医療機器(IVD)」ごとの分類ルールが記載されています。該当するルールを特定します。 - クラス(Class I〜IV)を決定
該当ルールに従ってリスクレベルを評価し、仮のクラスを決めます。 - 各国の分類基準と照合
米国ならFDAのProduct CodeやClassification Database、EUならMDR Annex VIIIを確認し、GHTFベースの分類が各国でどう位置付けられているかを照合します。
日本の場合はこちらを参照ください。
・クラスⅠ(一般医療機器):GHTFのクラスAに相当
・クラスⅡ(管理医療機器):GHTFのクラスBに相当
・クラスⅢ・Ⅳ(高度管理医療機器):GHTFのクラスC・Dに相当 - 不明確な場合は当局や専門家に相談
判断が難しい場合はPMDAの簡易相談、コンサルタント、規制当局のガイドライン等で確認します。
申請区分とは
医療機器を販売・製造するには、薬機法に基づく承認・認証・届出などの手続きを行う必要があります。このとき、機器のクラス分類(リスクレベル)だけでなく、製品のタイプ(新規・改良・後発)によっても「申請区分(承認区分)」が決まります。
申請区分
- 製造販売届出(主にクラスⅠ):低リスク機器で、届出だけで販売可能
- 認証(第三者認証)(主にクラスⅡ):指定基準に適合する場合は、認証機関で認証
- 承認(PMDA承認)(主にクラスⅡ〜Ⅳ):新規性が高い、または高リスクで基準がない場合はPMDAの審査・承認が必要
新規・改良・後発の違い
- 新規医療機器
国内外で類似の承認実績がない、新しい構造・原理・適応を持つ機器。リスク評価や基準が確立していないため、原則PMDA承認が必要になります。 - 改良医療機器
既存承認品に対し、構造・原理・適応・材質などの一部を改良した機器。改良内容によってはリスクレベルやクラス分類が変わる可能性があり、承認・認証・届出のいずれになるかは個別判断が必要です。 - 後発(ジェネリック)医療機器
既存承認品と同一の仕様・性能を持つ機器。多くの場合、基準に適合すれば第三者認証や届出で済みますが、適合基準がない場合は承認が必要になることもあります。
申請区分の調査・判定の進め方
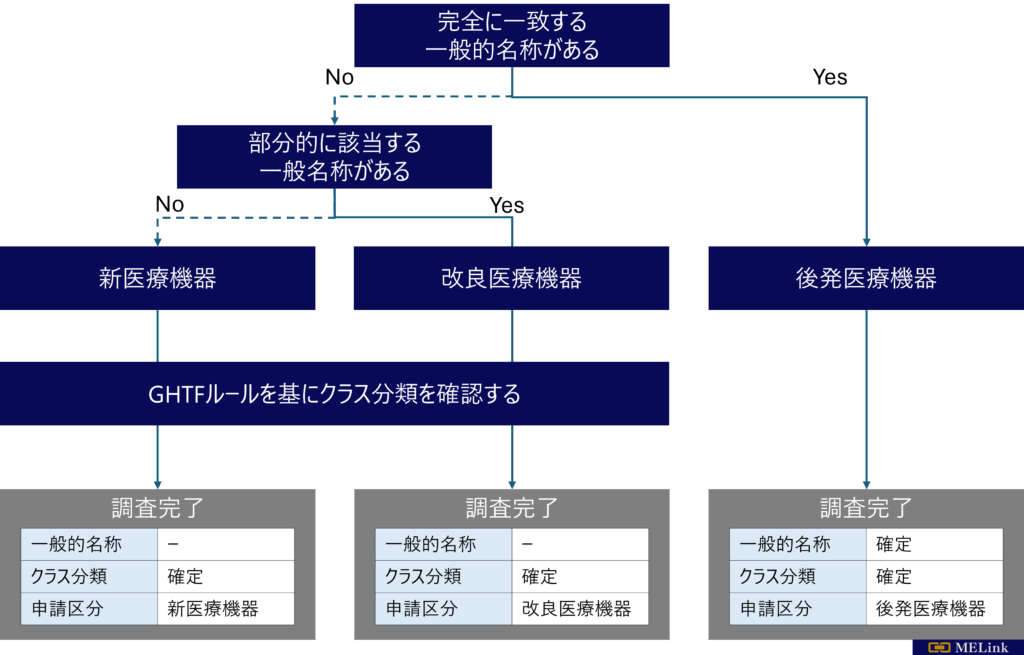
- 一般的名称を調査する
完全に一致する、一般的名称があるかを確認してください。ある場合は、後発医療機器の取り扱いとなります。
部分的に一致する一般的名称がある場合は、改良医療機器の取り扱いの可能性が高いです。
どの一般的名称にも該当しない場合は、新医療機器の取り扱いになる可能性が高いです。 - 同種同効の既承認機器を参考にする
仕様・性能・適応が近い機器の区分を確認し、自社製品が新規・改良・後発のどれに当たるか判断します。 - 判断が難しい場合は事前相談を活用
PMDAの簡易相談で、申請区分や必要試験の考え方を確認できます。
調査が終わったら
PMDAの簡易相談または開発前相談を活用して、自社の認識を確認してください。PMDA相談には準備面談と対面助言の2種類がありますが、対面助言をしておくと公式的な記録(議事録)が発行されるので、なおよいです。
PMDA相談の解説は別ページにていたします。